醫療器械法規注冊及計算機軟硬件零售領域的合規要點
隨著醫療科技的飛速發展,醫療器械與信息技術的融合日益加深。醫療器械的法規注冊,以及與之相關的計算機軟硬件及輔助設備的零售活動,構成了一個復雜而專業的交叉領域。本文旨在解讀其核心法規要求與常見問題,為相關從業者提供指引。
一、 醫療器械法規注冊的核心框架
醫療器械的注冊管理,核心目標是確保產品的安全、有效和質量可控。在中國,其主要依據是《醫療器械監督管理條例》。根據風險等級,醫療器械分為第一類(低風險)、第二類(中風險)和第三類(高風險),實行分類管理。
- 注冊/備案流程:
- 第一類醫療器械:實行產品備案管理,向所在地設區的市級藥品監督管理部門提交備案資料。
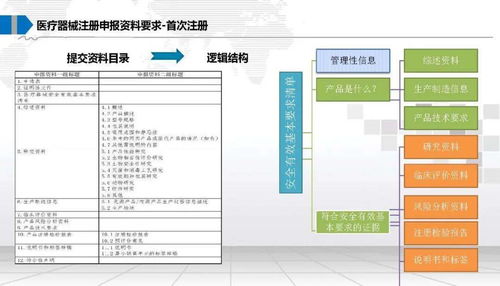
- 第二類、第三類醫療器械:實行產品注冊管理。申請人需向國家藥品監督管理局(NMPA)或指定的省級藥品監督管理部門提交包含產品研制報告、臨床評價資料、產品技術要求、風險分析報告等在內的全套注冊申請資料,經技術審評、行政審批通過后獲得醫療器械注冊證。
- 臨床評價要求:高風險產品通常需要進行臨床試驗,以驗證其安全有效性。部分低中風險產品可通過同品種比對等方式進行評價。
- 質量管理體系:醫療器械注冊申請人/注冊人必須建立符合《醫療器械生產質量管理規范》的質量管理體系,并在注冊過程中接受現場核查。
二、 與計算機軟硬件零售的交叉與合規要點
當醫療器械的功能實現高度依賴于特定的計算機、服務器、專用硬件或軟件時,這些軟硬件設備便不再是普通的電子產品,其流通和使用需考慮醫療器械法規的延伸監管。
- 作為醫療器械組成部分的軟硬件:
- 如果計算機或特定硬件是醫療器械整機不可分割的一部分(如內置在醫療設備中的控制計算機、圖像處理工作站),其安全性、有效性已包含在整機的醫療器械注冊證中。零售商銷售此類“整機”時,必須確認其已取得有效的醫療器械注冊證。
- 如果軟件本身被界定為獨立軟件醫療器械(如影像處理軟件、輔助診斷軟件),則無論以何種載體(光盤、U盤、在線下載)提供,該軟件本身必須完成醫療器械注冊或備案。零售商銷售此類軟件,實質上是在銷售醫療器械。
- 作為醫療器械輔助設備的軟硬件:
- 許多通用計算機、服務器、顯示器、網絡設備等,被用作已注冊醫療器械的顯示、存儲、傳輸終端。此類零售活動屬于普通的“計算機軟硬件及輔助設備零售”。
- 核心合規點在于:零售商需明確告知客戶,這些通用產品并非醫療器械,不能單獨用于醫療目的,其性能和配置需滿足其所連接醫療器械的制造商明確規定的技術要求。避免宣傳其具有醫療功能,以防構成未經注冊的醫療器械的虛假宣傳或非法銷售。
- 數據安全與隱私保護:用于處理醫療數據的計算機和存儲設備,需符合《網絡安全法》、《數據安全法》及《個人信息保護法》的要求,確保醫療健康數據的安全存儲與傳輸。零售商在提供解決方案時,應提示客戶相關法律義務。
三、 常見問題與風險提示
1. 問題:銷售用于醫療環境的通用電腦,需要醫療器械經營許可證嗎?
解讀:如果該電腦本身不具備獨立的醫療功能,未被定義為醫療器械,僅作為通用計算平臺,則其銷售不需要醫療器械經營資質。但應確保其質量可靠,并建議客戶遵循醫療器械制造商對配套設備的技術規范。
2. 問題:為醫院信息系統(HIS)提供服務器集成服務,涉及法規嗎?
解讀:需區分情況。如果HIS系統本身是取得注冊證的醫療器械軟件,那么為其提供定制化集成服務可能被視為醫療器械經營活動的一部分,需與系統注冊人明確責任劃分。如果僅為通用IT基礎設施部署,則不直接受醫療器械法規管轄,但需嚴格遵守醫療行業的數據安全和等保要求。
- 風險提示:最大的法律風險在于“模糊地帶”的違規。
- 非法銷售未經注冊的醫療器械:將具有醫療功能(如診斷、治療、監測)的軟件或軟硬件一體設備,作為普通商品銷售。
- 虛假宣傳:對通用的計算機硬件進行具有醫療效果的宣傳,誤導消費者。
- 質量責任:提供的輔助設備因質量問題導致連接的醫療器械數據錯誤或功能失效,可能需承擔相應的民事及產品質量責任。
四、 與建議
在醫療器械與信息技術深度融合的背景下,從事相關軟硬件零售和服務的企業,必須建立清晰的合規邊界意識:
- 準確分類:嚴格區分所售產品是“醫療器械”本身,還是“醫療器械的通用輔助工具”。
- 資質查驗:銷售醫療器械(包括獨立軟件),必須查驗并留存有效的醫療器械注冊證和供應商的醫療器械經營許可證。
- 宣傳合規:宣傳內容實事求是,不對非醫療器械產品明示或暗示醫療功能。
- 強化合同與告知:在銷售合同中明確產品屬性、用途限制,并向客戶提供必要的技術符合性說明與風險提示。
- 關注數據安全:主動了解和滿足醫療行業對信息安全的特殊要求。
通過建立完善的內部合規流程,相關企業不僅能有效規避法律風險,更能贏得醫療機構和患者的信任,在醫療健康產業數字化浪潮中行穩致遠。
如若轉載,請注明出處:http://www.aooqo.cn/product/66.html
更新時間:2026-04-16 12:35:36